J. C. A. Prentice, R. J. Charlton, A. A. Mostofi and P. D. Haynes, Combining embedded mean-field theory with linear-scaling density-functional theory, Journal of Chemical Theory and Computation 16, 354 (2020)
Quantum embedding methods are a vital tool in the study of complex molecular or extended systems, where the most interesting physics or chemistry is associated with a particular region, but the rest of the system still interacts significantly with this “active region”, in a way that can only be described using quantum mechanics. Using an accurate quantum mechanical method (e.g., hybrid density functional theory (DFT)) may be necessary to correctly predict the properties of the active region, but using this level of theory for the entire system may be prohibitively expensive. Instead, quantum embedding schemes enable the active region to be treated at a high level of theory, whilst simultaneously treating the environment at a lower, less computationally demanding level of theory (e.g. semi-local DFT). The result is a calculation that has the accuracy of the high-level theory, but at a fraction of the cost.
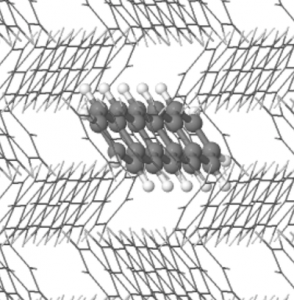
In a paper published in the Journal of Chemical Theory and Computation, we present a novel implementation of embedded mean field theory (EMFT) into the linear-scaling DFT code ONETEP, enabling efficient hybrid DFT-in-semi-local DFT quantum embedding calculations on large-scale systems. We have applied our implementation to a range of systems, including pentacene, pentacene-doped crystalline p-terphenyl, and metallocene-doped carbon nanotubes, and show it provides excellent results compared to full system hybrid DFT calculations and experiment. In particular, in pentacene-doped p-terphenyl, the ability to include a much larger p-terphenyl environment than previous calculations, whilst still using hybrid DFT for the pentacene dopant itself, gives much better agreement with experimental measurements of the shift in the S0 to S1 excitation due to the presence of the p-terphenyl host. This calculation contained nearly 3000 atoms, an order of magnitude larger than any calculation attempted before with EMFT.
This work was done as part of Dr Joseph Prentice’s postdoctoral research funded by the EPSRC (grant no. EP/P02209X/1) and Robert Charlton’s PhD in the Centre for Doctoral Training in Theory and Simulation of Materials. The project was supervised by Peter Haynes and Arash Mostofi.