N. V. Tepliakov, R. Ma, J. Lischner, E. Kaxiras, A. A. Mostofi and M. Pizzochero, Dirac half-semimetallicity and antiferromagnetism in graphene nanoribbon/hexagonal boron nitride heterojunctions, Nano Letters 23, 6698 (2023).
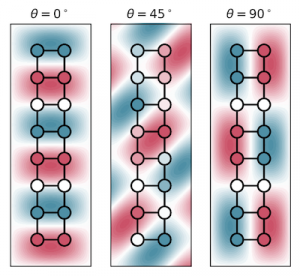
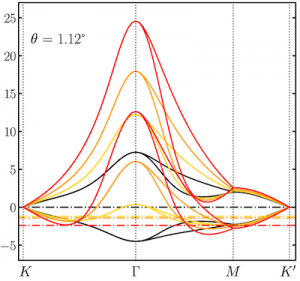
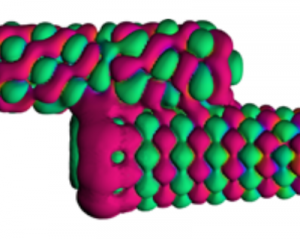
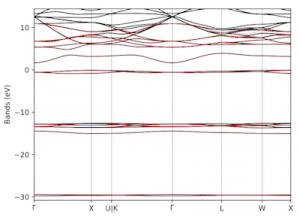
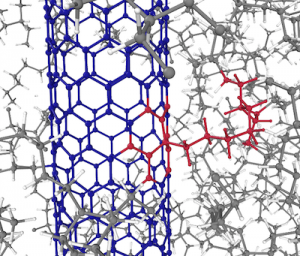
Spintronics is an emerging branch of electronics that aims to harness the spin of electrons, a purely quantum mechanical feature, for storing and processing information. The development of spintronic devices relies on discovering materials in which electrons with opposite spin orientations behave differently. In our recent work, carried out in collaboration with researchers at Harvard University, we examined graphene nanoribbons, one-dimensional strips of carbon atoms, embedded in hexagonal boron nitride, a two-dimensional insulator. Using advanced computer simulations, we revealed that this heterojunction is half-metallic, that is, electrons exhibit conducting behaviour for electrons of one spin orientation and insulating behaviour for those with the opposite spin orientation. Further, we found that charge doping the heterojunction can drive a transition between two different magnetic states of the nanoribbon (an antiferromagnetic and a ferrimagnetic one). Our results show that heterojunctions comprising graphene nanoribbons embedded in hexagonal boron nitride are a promising platform for future spintronic applications thanks to their combination of half-metallic behaviour and electrically tunable magnetism.
This work was part of the PhD work of Nikita Tepliakov and was in collaboration with colleagues Michele Pizzochero and Efthimios Kaxiras at Harvard University.